

A quality management system is the backbone of the medical device manufacturing facility. The QA experts are involved from day one of the product development cycle to mitigate risk and improve processes. The dynamic medical device manufacturing trends are taking a major leap in 2021. Medical devices are much more complex in terms of manufacturing and easier in terms of operation. It’s a transition phase and quality assurance need an upgrade as well.
The data is a billion-dollar industry and healthcare devices shall utilize its power too. Some of the existing quality regulations are outdated and do not address the cyber threat and data integrity-related issues of the medical devices. The role and responsibilities of Quality Assurance are not static; these evolve with time. The core aim of Quality Assurance management system is to ensure risk mitigation for all the stakeholders involved in the processes continuously.
How to determine when to establish a QMS?
Usually, there is no set formula for setting a QMS in medical device manufacturing. Many manufacturers do not implement the QMS process in the beginning and that seems like a cakewalk initially but later it becomes a bone of contention. The later stages require documentation to show that they had controls in place since the beginning of the manufacturing. The DHF (Design history file) and technical file are the prime examples of the same. The design history record, SOP, change control, and risk management is necessary for the device regulatory submission dossier. Thus, it’s recommended to have a QMS established since day one of the product device manufacturing cycle.
Let’s see how does quality assurance work for continuous upgrade of the existing processes for enhanced outputs.
The PDCA lifecycle of the medical device manufacturing
- Plan: Quality Planning
- Do: Quality Control (QC)
- Check: Quality Assurance (QA)
- Act: Quality Improvement
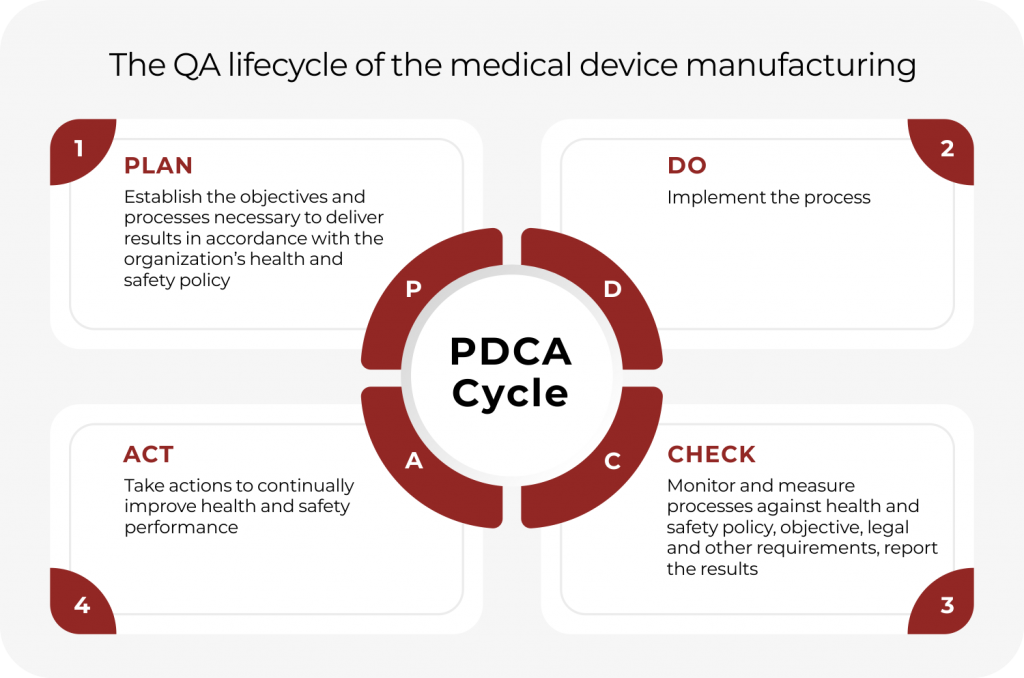
The PDCA cycle is the easier way to understand how the system of quality assurance operates in the manufacturing facility at various levels. The PDCA cycle is Plan, Do, Check, and Act.
Plan
Evaluating the existing procedures based on set industry standards or with a more advanced reference is the primary motive of this stage. The planning here helps in setting up strategies to implement ahead for improving the existing procedures for enhanced results. The predictive analysis at this stage is important for proposing changes that improve the process and maintain traceability.
To summarize,
- Identification of risks & opportunities within the QMS
- Planning resources & identifying competencies
- Raising awareness
- Setting up communication
- Determining & implementing the support structure.
Do
The second stage is the implementation stage where all the changes identified in the prior phase of evaluation are put into practice.
Check
The check stage involves rigorous evaluation and testing of the changes. If the visible outcomes of the process are in a positive direction, then the changes made are sustainable and these can run over a long time. In several cases, the changes might not be beneficial. Such changes need instantaneous deletion from the system or maybe re-evaluation with enhancements. Sometimes the result of the changes proposed might vary with the expected “Plan” stage. In such cases, the PDCA cycle can run several times to compare the results and feasibility of the changes proposed.
Act
This stage involves action on the changes proposed in the facility. After the checks have been made and all the evaluations are done, finally, action can be taken to implement the changes for cost optimization, improving efficiency, and building a win-win situation for both companies and its customers/clients.
Significance of ISO 13485 in medical device manufacturing
ISO 13485 is the set of regulations marking the credibility of medical devices being produced in any facility. While complying with regulations set by ISO 13485, one mitigates the risks associated with all the stakeholders, patients, users, customers, and regulatory agencies. It’s not just a set of rules to market the devices, sticking to guidelines set by the ISO 13485 reduces the failures, surprises, and risks associated with the manufacturing processes as well as at the later marketing stage. Compliance with ISO 13485 mandates patient safety and the manufacturer’s reputation. As technology is becoming a crucial part of medical devices ISO 13485 has revised its clauses to mitigate risks in future medical devices using high-end software and data integration. Clauses 4.1.6, 7.5.6, and 7.6 under the revised ISO 13485 address the risks associated with validation and re-validation of the software in the QMS processes.
Some key benefits of having QMS in medical manufacturing include:
- Reduced market time for devices
- Deep root cause analysis in case of any error
- Adaptability in compliance with industry regulations
- Eradication of non-value added and waste activities
- Improved supplier relations and management
QMS for future medical devices
Although, we have enough guidelines to check and evaluate the current processes. The upcoming challenges in the future medical devices remains a tough net to crack. Let’s see FDA’s planning about mitigating risks associated with the future medical devices.
FDA’s steps to mitigate risks in future
FDA plans to set up an entirely new arena for mitigating the risks and threats associated with future medical devices. Here are the major steps which FDA plans for assuring quality in future medical devices.
-
Addressing the cybersecurity concerns in future medical devices
Substantial integration of medical devices to the cloud has risen cybersecurity concerns. A hacker might break into the system putting sensitive information at risk. FDA is considering a new pre-market regulation where companies will be required to incorporate device-oriented data safety measures. Manufacturers would also be required to submit a “software bill of material” along with product submission to make authentic information about the product available to customers.
-
Promoting sustainable innovation
Everyone in the industry is awestruck by the new generation of technologies. Their vast potential is being deployed in every possible way. Manufacturing automation and device upgrades in terms of operation, accessibility, and safety are few primary offerings of the new-gen technologies. But like it’s said everything that shines aren’t gold!
The app integration and cloud sharing of data over interconnected medical devices have made things complicated. The sustainability of the devices is directly proportional to the data integrity they can offer. FDA is devising ways to create streamlined pathways for comparative safety claims to promote the sustainability of these medical devices in the future.
-
Product-based lifecycle approach
FDA’s medical device center, CDRH is organized according to the stage of the product lifecycle. The regulations, compliance, pre-market and post-market surveillance are currently dependent on the product lifecycle rather than the product being regulated. In the future FDA plans to re-structure the compliance according to the type of product being regulated.
Conclusion
The role of Quality Assurance is to check processes and procedures in compliance with the ISO13485: 2016, USFDA and MDSAP standards. Implementing a full QMS ensures quality outputs maintaining medical device design, manufacturing, risk management, supplier management, product labeling, storage & distribution, clinical data, and complaint handling. The role of QMS cannot be undermined at any level. Also, upcoming trends in future medical devices shall advance the Quality management systems for digital congruency to develop advanced Class I, Class II, and Class III medical devices.